- Ayrıntılar
- Editör tarafından yazıldı.
- Kategori: Ders Notu Özeti
- Gösterim: 18500
NEOPLAZİ - ÖZET NOT
Beniğn yada maliğn her tümör iki ana komponentten oluşur.
1- Parankim: Çoğalan tümöral hücreler
2- Stroma: Bağ dokusu ve kan damarlarından oluşan destek doku
Belirgin kollajenöz stroma oluşumu dezmoplazi, yoğun stroma içeren tümörler skiröz olarak isimlendirilir. Karsinomlar sarkomlara kıyasla daha dezmoplazik, yani daha skiröz tümörlerdir. Örneğin: İnvaziv duktal meme karsinomu
İsimlendirme
Beniğn Tümörler:
Genellikle köken aldıkları hücre tipinin sonuna –oma eki alarak isimlendirilirler.
Yumuşak doku (mezenşimal) tümörlerinden fibroma, kondroma, osteoma gibi...
Epitelyal beniğn tümörler ise hücre tipi ve oluşturdukları yapıya göre isimlendirilir.
• Adenom : Glandüler yapılar oluşturan veya gland kökenli beniğn tümör.
• Papillom : Mikroskobik veya makroskobik parmaksı çıkıntılar yapan epitelyal beniğn tümör.
• Kistadenom – papiller kistadenom : Özellikle over ve apendikste epitel hücreleri ile döşeli, bazen lümene papiller çıkıntılar oluşturabilen kist oluşumu ile karakterli beniğn tümörler.
• Polip : Mide ve kolon gibi boşluklu organ-larda, mukozadan kabarık saplı veya sapsız (sesil) beniğn tümörler.
Maliğn tümörler (kanser):
• Mezenşimal kökenli maliğn tümörler, köken aldığı hücre tipinin sonuna sarkom eki alarak isimlendirilir. Liposarkom, Fibrosarkom, Kondrosarkom, Leiomyo-sarkom, Rabdomyosarkom vb.
• Epitelial kökenli maliğn tümörler ise köken aldığı hücre tipinin sonuna karsinom eki alırlar. Adenokarsinom, Skuamöz hücreli (yassı epitel hücreli) karsinom, transisyonel hücreli (değişici epitel hücreli) karsinom vb.
Beniğn gibi isimlendiren maliğn tümörler:
Seminoma, hepatoma, lenfoma, mezotelioma, kordoma, melanoma, hipernefroma (renal hücreli karsinom)
Mikst tümörler: Tek bir hücreden köken aldığı halde hem epitelial hemde mezenşimal diferansiasyon gösteren tümörlerdir.
- Pleomorfik adenom
- Fibroadenom
- Wilms tümörü
- Sinovial sarkom
- Maliğn mezotelioma
- Uterusun mikst müllerien (mezodermal) tümörleri (karsinosarkom)
-Memenin filloides tümörü
Teratom : Üç germ yaprağından köken alan germ hücre tümörüdür.
Koristom : (Heterotopi) Normal bir doku örneğinin olması gerekenden farklı bir yerde bulunmasıdır. Mide mukozasında pankreas dokusu veya böbrek parankiminde adrenal dokusunun varlığı, Mekel divertikülünde mide mukozasının varlığı gibi..
Hamartom : Normalde belirli bir dokuyu oluşturan matür hücrelerin, gelişim defekti sonucu, aynı yerde düzensiz ve disorganize kitle oluşturmalarıdır. Klinikte kitle oluşturarak tümörle karışabilir. Mikroskobik olarak ayırımı kolaydır. Akciğerde en sık görülen beniğn lezyon hamartomdur.
Beniğn – Maliğn Tümör Ayırımı
Diferansiasyon ve anaplazi, büyüme hızı, lokal invazyon ve metastaz ayırımda kullanılan kriterlerdir. En kesin maliğnite göstergesi metastazdır.
• Diferansiasyon : Tümörün köken aldığı hücreleri taklit edebilme yeteneğidir. İyi diferansiye tümörler düşük dereceli (grade’li), köken aldığı hücrenin özelliklerini taşıyan tümörlerdir ve mikroskobik incelemede kolay tanınırlar. Endokrin kökenli iyi diferansiye tümörler, hücrenin fonksiyonunuda taklit ederek hormon üretebilirler. Tümörün diferansiasyonu azaldıkça (az diferansiye, indiferansiye tümör) derecesi artar, prognozu bozulur ve tanısı zorlaşır.
• Anaplazi : Diferansiasyonun tamamen kaybıdır. Özellikleri:
- Pleomorfizm: Hücre ve hücre nüvesinde, boyut, şekil ve boyanma farklılığı
- Anormal nükleer görünüm: DNA içeriği artmış, hiperkromatik ve iri nüveler, belirgin nükleol varlığı
- Mitoz: Artmış tipik ve atipik (tripolar, quadripolar) mitozlar
- Polarite kaybı: Oryantasyon bozukluğu, polarite kaybı, anarşik, disorganize büyüme
- Diğerleri: Tümör dev hücreleri, tümör merkezinde iskemik nekroz
• Displazi: Bazal membranı aşmayan, intraepitelial gelişen, hücrelerde üniformite kaybı ve yapısal düzenlemede bozulma…Displastik değişiklikler belirgin olup, epitelin tüm kalınlığını içine alıyorsa: karsinoma in situ.. Hafif ve orta dereceli displazilerde, etken ortadan kaldırılırsa, epitel çoğu kez normale döner.
• Büyüme Hızı: Maliğn tümörler beniğn tümörlere göre daha hızlı büyümekle birlikte istisnalar olabilir. Örneğin hamilelikte östrojen artışına bağlı olarak leiomyom hızla büyüyebilir.
• Lokal İnvazyon: Metastazdan sonra malğnite tanısında ikinci önemli kriterdir. Beniğn tümörlerin çoğu kapsüllü olup çevre dokudan kolay ayrılırlar. (Kapiller hemanjiom ve pleomorfik adenom, beniğn oldukları halde halde invazivyon yapabilirler.) Maliğn tümörlerde ise çevre dokuya invazyon belirgindir ve operasyonda bir miktar normal çevre doku ile birlikte çıkarılmalıdır (radikal cerrahi).
• Metastaz : En kesin maliğnite kriteri olup primer tümörle devamlılığı olmayan tümör implantlarıdır. (Glial tümörler ve bazal hücreli karsinom maliğn olduğu halde uzak metastazları çok nadirdir.) Metastaz üç şekilde gerçekleşebilir.
Beniğn-Maliğn Tümör Ayırımı
| Özellikleri | Beniğn | Maliğn |
| Diferansiasyon/anaplazi | İyi diferansiye, köken aldığı dokuya benzer | Diferansiasyon kaybı; anaplazi, yapısal atipi |
| Büyüme hızı | Genellikle yavaş, büyüme durabilir, hatta gerileyebilir; mitotik figürler nadir ve tipik | Yavaş yada hızlı olabilir; mitotik figürler sık ve atipik |
| Lokal invazyon | Genellikle belirgin sınırlı; invazyon yok | Lokal invaziv, çevre dokuları infiltre eder; bazen sınırlı görülür |
| Metastaz | Yok | Genellikle var; primer ne kadar büyük ve az diferansiye ise, metastaz o kadar sıktır |
Epidemiyoloji
Kanser Sıklığı
Organ tümörleri içerisinde, erkekde en sık görülen tümörler prostat, akciğer ve kolorektal karsinomlardır. Kadında meme, akciğer, kolon ve rektum karsinomları sıktır. Akciğer karsinomu, her iki cinste, en sık ölüme yol açan tümördür.
Coğrafi ve Çevresel Faktörler
Japonya’da mide karsinomu; ABD ve Belçika’ da akciğer karsinomu sıktır. Yeni Zelanda’ da maliğn melanom; İzlanda’ya göre 6 kat fazladır.
Kişisel alışkanlıklar, çalışma ortamında maruz kalınan etkenler, besin maddeleri gibi pek çok çevresel etken kansere yol açabilir. Obesite risk faktörüdür.
Alkol : Orofarinks, larinks, özefagus, karaciğer kanserleri
Sigara : Ağız, farinks, özefagus, mide, pankreas, mesane, böbrek, serviks ve en önemlisi akciğer kanserleri
Alkol ve Sigara : Üst solunum – sindirim sistemi kanserleri riskini, birlikte kullanılınca, tek başına etkilerinden daha fazla arttırırlar.
Çalışma Ortamındaki Karsinojenler
| Etken | Kanser tipi | Kullanım alanı |
| Arsenik | Akciğer, cilt, hemanjiosarkom | Metal eritme ürünü, elektrikli aletler, ilaçlar, herbisid ve fungusidler |
| Asbest | Akciğer, mezotelioma, GİS (özefagus, mide, kalın barsak) | Ateşe dayanıklı kumaşlar, döşeme ve duvar kağıtları, eskiden gemi yapımı |
| Benzen | Lösemi, Hodgkin lenfoma | Lamba yağları, baskı, boya, kuru temizleme, yapıştırıcı, deterjanlar |
| Berilyum | Akciğer | Yakıt ve uzay araçları, nükleer reaktörler |
| Kadmiyum | Prostat | Sarı pigment, fosfor, piller, metal kaplama |
| Krom | Akciğer | Metaller, boyalar, pigment ve koruyucular |
| Etilen oksid | Lösemi | Meyve ve kuruyemişlerin olgunlaştırılması, roketin fırlatılması, kimyasal sentez, gıda ve tekstilde ilaçlama, sterilizanlar |
| Nikel | Burun, akciğer | Nikel kaplama, seramik ve pil ürünleri |
| Radon | Akciğer | Uranyum içeren minerallerin çürümesi, madenler |
| Vinil klorid | Anjiosarkom, karaciğer | Dondurucu, plastikler için yapıştırıcı; eskiden aerosollerde bulunurdu, |
Yaş
Kanserlerin çoğu 55 yaş üzerinde görülür. 60-79 yaş arasında, kansere bağlı ölümlerde pik oluşur. 15 yaş altı çocuklarda kanserden ölüm oranı %10 olup, ABD’de kazalardan sonra ikinci ölüm nedenidir. Akut lösemi ve SSS tümörleri, bu ölümlerin %60’ını oluşturur.
OD herediter kanser sendromları
• Familyal retinoblastom: Genellikle bilateral, osteojenik sarkom gibi ikincil tümörler
• Familyal adenomatöz polipozis koli: Sayısız polipöz adenomlar, profilaktik kolektomi yapılmazsa 50 yaş civarında %100 kanser gelişimi
• MEN tip I: Pituiter, paratiroid, pankreas ada hücreleri adenomları
• MEN tip II: Tiroid meduller karsinomu, feokromasitoma, paratiroid tümörleri
• MEN tip III: Tiroid meduller karsinomu, feokromasitoma, ganglionöromlar
• Nörofibromatozis tip I (von Recklinghausen hastalığı) : Multiple nöral tümörler (beyin ve optik sinir gliomları, feokromasitoma, nörofibrom, schwannom, menenjiom), café au lait lekeleri , pigmentli iris hamartomu (Lisch nodülleri), kemik defektleri ve maliğn periferik sinir kılıfı tümörü (MPSKT) riski
• Nörofibromatozis tip II: Bilateral akustik sinir tümörleri (schwannom, menenjiom) cilt tümörleri, café au lait lekeleri.. Lisch nodülleri görülmez. MPSKT riski var.
• Von Hippel-Lindau sendromu: Retina, serebellum, beyin sapı hemanjiomları, organ kistleri ve renal hücreli karsinom riski
• Wilms tümörü : Konjenital malformasyon ve böbrekte Wilms tümörü
• Li Fraumeni Sendromu: Meme, kolon kanserleri, sarkomlar gibi çok sayıda tümörler
• Herediter nonpolipoid kolon kanser (HNPCC): DNA tamir defekti…En sık görülen kanser predispozisyon sendromudur. Kolon, ince barsak, endometrium ve over tümörleri görülebilir.
OR herediter preneoplazik durumlar
• Kseroderma pigmentozum: Ciltte bazal hücreli ve skuamöz hücreli karsinom, melanom
• Bloom sendromu: Akut lösemi ve değişik karsinomlar
• Fankoni anemisi: Akut lösemi, skuamöz hücreli karsinom ve karaciğer kanser
• Ataksi-telenjiektazi: Akut lösemi, lenfoma, meme kanseri
X’e bağlı immün yetmezlikler
• X’e bağlı agamaglobulinemi: Lenfoma ve lösemi
• Wiskott-Aldrich sendromu: Akut lösemi, lenfoma
• X’e bağlı lenfoproliferatif sendrom: EBV’e anormal yanıt ve buna bağlı B hücreli immünoblastik lenfoma
Edinsel preneoplastik durumlar:
Rejeneratif, hiperplazik, metaplazik ve displazik durumlar tümöre yatkınlık oluşturursa da tümör riski oldukça düşüktür. Kronik atrofik gastrit, ciltte solar keratoz, ülseratif kolit, oral kavite – vulva – vajen ve penisde lökoplaki ve eritroplaki, villöz adenom, endometrial hiperplazi, skuamöz metaplazi, siroz gibi durumlar örnek verilebilir.
Ülseratif kolit, Crohn hastalığı, Helikobakter pylori gastriti, viral hepatit ve kronik pankratit gibi kronik inflamatuar olaylarda kanser riski artar. Bunun nedeni tam olarak bilinmese de, sitokinlerin trasforme hücrelerin çoğalmasını uyardığı düşünülmektedir. İnflamasyonda oluşan serbest radikaller mutajenik olabilir. Kolon kanseri gibi bazı tümörlerde COX-2 artışı belirlenmiş olup tedavide COX-2 inhibitörlerinin rolü araştırılmaktadır.
Kalıtsal Kanser Predispozisyonu
Otozomal dominant
| Gen | Predispozisyon |
| RB | Retinoblastom |
| p53 | Li-fraumeni sendromu |
| p16INK4A | Melanom |
| APC | FAP/kolon kanser |
| NF1, NF2 | Nörofibromatozis 1 ve 2 |
| BRCA1, BRCA2 | Meme ve over tümörleri |
| MEN1, RET | MEN 1 ve 2 |
| MSH2, MLH1, MSH6 | Herediter nonpolipozis kolon kanser |
| PATCH | Nevoid bazal hücreli karsinom |
| Otozomal Resesif | |
| Kseroderma pigmentozum | |
| Ataksi telenjiektazi | |
| Bloom sendromu | |
| Fankoni anemisi |
Kanserin Oluşumu
Dört grup normal düzenleyici gende oluşan mutasyon kanser gelişiminde önemlidir: Çoğalmaya yol açan protoonkogenler; çoğalmayı durduran tümör süpresör genler; apoptoz genleri ve DNA tamir genleri…
Protoonkogen mutasyonu (onkogen) ve ilişkili olduğu tümörler
|
Kategori |
Protoonkogen |
Aktivasyon şekli |
Tümör |
|
Büyüme faktörleri |
|||
|
PDGF |
SIS |
Overekspresyon |
Astrositom |
|
FGF |
HST-1 |
Overekspresyon |
Mide, mesane, meme karsinomları, melanom |
|
INT-1 |
Amplifikasyon |
||
|
TGF-alfa |
TGF-alfa |
Overekspresyon |
Astrositom,hepatoselüler karsinom |
|
HGF |
HGF |
Overekspresyon |
Tiroid karsinomu |
|
Büyüme faktör reseptörleri |
|||
|
EGF-resptör ailesi |
ERB-B1 |
Overekspresyon |
Akciğerde skuamöz karsinom, gliom |
|
CSF-1 reseptörü |
FMS |
Nokta mutasyonu |
Lösemi |
|
Nörotropik faktör reseptörü |
RET |
Nokta mutasyonu |
MEN 2AveB, ailesel medüller tiroid karsinomu |
|
PDGF reseptörü |
PDGF-R |
overekspresyon |
Gliomlar |
|
Kök hücre faktör reseptörü |
KIT |
Nokta mutasyonu |
Gastrointestinal stromal tümörler ve diğer yumuşak doku tümörleri |
|
Sinyal taşıyıcı proteinler |
|||
|
GTP-bağlama |
K-RAS |
Nokta mutasyonu |
Kolon, akciğer, pankreas tümörleri |
|
Nonreseptör tirozin kinaz |
ABL |
Translokasyon |
KML, ALL |
|
RAS sinyal iletimi |
BRAF |
Nokta mutasyonu |
Melanom |
|
WNT sinyal iletimi |
Beta-katenin |
Nokta mutasyonu Overekspresyon |
Hepatoblastom, hepatoselüler karsinom |
|
Nükleer düzenleyici proteinler |
|||
|
Transkripsiyon aktivatörleri |
C-MYC |
Translokasyon |
Burkitt lenfoma |
|
Hücre siklus düzenleyicileri |
|||
|
Siklinler |
CYCLIN D |
Translokasyon |
Mantle zon lenfoma |
|
Sikline bağlı kinazlar |
CDK4 |
Amplifikasyon yada nokta mutasyonu |
Glioblastom, melanom, sarkom |
Kromozomal translokasyonlar, onkogenleri aktive ederler. Lösemi ve lenfomaların gelişimindeki temel mekanizma budur.
Onkogen Translokasyonu
| Maliğnite | Translokasyon | Etkilenen genler |
| Kronik myeloid lösemi | (9;22) | Abl(9), bcr (22) |
| AML ve ALL | (4;11) | AF4 (4), MLL (11) |
| Burkitt lenfoma | (8;14) | c-myc (8), IgH (14) |
| Mantle hücreli lenfoma | -11,14 | Cyclin D (11), IgH (14) |
| Foliküler lenfoma | (14;18) | IgH (14), bcl-2 (18) |
| T hücreli ALL | (8;14) | c-myc (8), TCR-alfa (14) |
| Ewing sarkom | (11;22) | FI-1 (11), EWS (22) |
Onkogenler ve Tedavi
• ERB-B2 (HER2 neu) mutasyonu, yalnızca tümör hücrelerinde olduğundan, buna yönelik antikorlar (transtuzumab) tedavide kullanılmaktadır. Buna hedeflenmiş tedavi denir.
• Diğer başarılı bir tedavi yöntemi ise, gastrointestinal stromal tümörlerde, c-KIT’in, reseptör tirozin kinaz aktivitesinin, imatinib ile bloke edilmesidir.
• KML ve bazı ALL olgularında, tirozin kinaz aktivitesi içeren protoonkogen c-ABL, 9. kromozomdaki normal yerinden koparak 22. kromozoma geçer ve BCR geni ile birleşir. Bu füzyon ile tirozin kinaz aktivitesi artar. İmatinib mezilat, BCR-ABL tirozin kinazı hedefleyerek tedavi sağlar. Toksisitesi düşüktür.
Tümör süpresör genler ve ilişkili oldukları tümörler
| Subselüler lokalizasyon | Gen | Fonksiyon | Somatik mutasyonlu tümörler | Kalıtsal mutasyonlu tümörler |
| Hücre yüzeyi | TGF-beta reseptörü
E-cadherin |
Büyüme inhibisyonu
Adezyon |
Kolon karsinomu
Mide karsinomu |
Bilinmiyor
Ailesel mide ca. |
| Plazma membran iç yüzeyi | NF-1 | RAS sinyali ve p21 inhibisyonu | Nöroblastom | Nörofibromatozis 1 ve sarkomlar |
| Hücre iskeleti | NF-2 | İskelet stabilitesi | Schwannom ve menenjiomlar | Nörofibromatozis 2, akustik schwannom ve menenjiomlar |
| Sitozol | APC/beta katenin | Sinyal iletimi inhibisyonu | Mide, kolon, pankreas ca. | FAP/kolon ca. |
| PTEN | PI-3 kinaz sinyal iletimi | Endometrium ve prostat ca | Bilinmiyor | |
| SMAD2 ve SMAD4 | TGF-beta sinyal iletimi | Kolon, pankreas tm | Bilinmiyor | |
| Nüve | RB | Hücre siklus düzenleyicisi | Retinoblastom, osteosarkom, meme, akciğer, kolon ca | Retinoblastom, osteosarkom |
| P53 | Hücre siklus arresti ve apoptoz | Çoğu tümörler | Li-Fraumeni sendromu, multiple karsinom ve sarkomlar | |
| WT-1 | Nükleer transkripsiyon | Wilms tümörü | Wilms tümörü | |
| P16 (INK4a) | Siklin bağımlı kinaz inhibisyonu | Pankreas, meme, özefagus karsinomları | Maliğn melanom | |
| BRCA-1 ve BRCA-2 | DNA tamiri | Bilinmiyor | Kadın meme ve over karsinomu; erkek meme karsinomu | |
| KLF6 | Transkripsiyon faktörü | Prostat | Bilinmiyor |
Apoptoz Genleri
• BCL-2 mutasyonu ile foliküler lenfoma gelişir. Foliküler lenfomaların %80’inde t(14;18) translokasyonu ile, 18. kromozomda yerleşen BCL-2, 14. kromozomdaki immünglobülin ağır zincir bölgesine geçer ve overeksprese olur. Apoptoz engellenir. B lenfositleri birikir ve yavaş çoğalan foliküler lenfomalar oluşur.
• MYC mutasyonu hücre çoğalmasını arttırarak, BCL-2 hücre ölümünü azaltarak tümör gelişiminde işbirliği yapabilirler.
• P53 mutasyonu varsa, BAX geni uyarılamaz ve apoptoz engellenir. Tümör riski artar. ¬
DNA tamir defektleri
• Herediter Nonpolipozis kanser sendromu: Çekum ve proksimal kolonda yerleşen tümörler ile karakterli, OD geçen bir kanser sendromudur. DNA eşleşme hataları oluşur. Örneğin, A ve T yerine, G ile T birleşir. Yanlış eşleşme tamir genlerinde defekt vardır. Protoonkogen ve tümör süpresör genlerde hatalı çiftlerin birikimi ile tümör riski artar. Endometrium ve over kanseride görülebilir.
• Kseroderma pigmentozum: Güneş ışınlerındaki UV etkisi ile cilt tümörleri gelişir. Primidin rezidülerinde çapraz bağlanma sonucunda DNA replikasyonu bozulur. Nükleotid eksizyon tamirinde defekt olduğu için DNA hasarı giderilemez.
• Diğerleri: Ataksi telenjiektazi ve Bloom sendromunda ionize radyasyona; Fankoni anemisinde ise DNA’yı çapraz bağlayan etkenlere karşı aşırı duyarlılık vardır. Ataksi telenjiektazi’de Purkinje hücrelerinin ilerleyici kaybı ile serebellar ataksi, defektif lenfosit proliferasyon ve matürasyonu ile immün yetmezlik, lenfoid maliğniteler, ionize radyasyona akut duyarlılık gelişir. ATM gen mutasyonu vardır. ATM, DNA hasarı oluştuğunda p53’ü uyarır. ATM eksikse, bu oluşmaz ve DNA tamiri ya da apoptoz başlatılamaz.
• BRCA-1 ve BRCA-2 genleri: BRCA-1 gen mutasyonu ile meme, over kanserleri yanı sıra prostat ve kolon kanseri riskinde hafif artış olur. BRCA-2 gen mutasyonu ise over ve erkek meme kanseri yanı sıra, melanom ve pankratik tümör riskini arttırır. Ailesel meme karsinomlarının çoğunda, bu genlerin mutasyonu belirlenirse de, diğer genlerden farklı olarak, sporadik olgularda etkileri gösterilememiştir.
Tümörde Anjiogenez
Tümörler beslenebilmek ve metastaz yapabilmek için damar oluşumunu uyarır (anjiogenez). Anjiogenez olmadan, 1-2 mm’den fazla büyüyemezler. İki önemli anjiogenik faktör; VEGF ve bFGF’ dir.
METASTAZ
Tümörün Yayılımı
Tümör büyümesi ve yayılımı dört aşamada gerçekleşir.
1-Transformasyon
2-Transforme hücrelerin çoğalması
3-Lokal invazyon
4-Metastaz
Çoğalan tümör hücreleri, bir taraftan yeni mutasyonlar geçirerek farklı subklonlar oluştururlar. Metastatik özellikler içeren subklon ne kadar erken gelişirse metastaz o kadar erken olur. Metastazın gelişmesinde bazı faktörlerin rolü vardır. Önce tümör hücreleri epitelial E-Cadherin gibi tutunma moleküllerini azaltarak ana kitleden koparlar. Daha sonra integrin reseptörleri ile laminin ve fibronektine tutunarak bağ dokusuna geçerler. Tümör hücreleri ve stroma hücrelerinden salgılanan proteazlar ile bazal membran ve bağ dokusu sindirilerek hücrelerin yolu açılır. Bunlar serin, sistein ve matriks metalloproteinaz (MMP)grubu proteazlardır. Sistein grubu proteazlardan katepsin D yüksekliğinin meme karsinomunda kötü prognoz bulgusu olduğu ileri sürülmektedir.
Damar endotelini aşarak dolaşıma giren tümör hücrelerine karşı ilk savaşan doğal öldürücü (NK) hücrelerdir. Trombositler ise tümör hücrelerinin yüzeyini örterek NK hücrelerinden korurlar Daha sonra T lenfositleri de devreye girerek tümöre karşı savaşır. Özellikle virüs etkisi ile oluşan tümörlerde sitotoksik T lenfositleri ön plandadır. Yerleşeceği bölgeye gelince endotel hücresine tutunan tümör hücreleri dışarı çıkarak integrin reseptörleri ve proteazların yardımı ile yerleşerek çoğalmaya devam eder. Tümör hücrelerindeki adezyon moleküllerinden CD44 (normalde T lenfositlerinde bulunur), tümör hücrelerinin ekstravazasyon ve yerleşmesinde rol oynar.
Hangi tümörün nereye metastaz yapacağı önceden kesin olarak belirlenemese de prostat karsinomu ön planda vertebraya; akciğer karsinomu adrenal gland, kemik ve beyine; nöroblastom karaciğer ve kemiğe metastaz yapar. Tümör hücreleri, hedef organ endoteline tutunabilen adezyon molekülleri içerebilir. Hedef organ, tümör hücreleri için kemotaktik olan, insülin benzeri büyüme faktörü I ve II üretebilir. Antiproteaz içeren çizgili kas dokusuna tümör hücrelerinin yerleşmesi zordur . Bu nedenle çizgili kasa metastaz nadirdir. Avasküler olan kornea ve kıkırdak gibi dokulara uzak metastaz oluşmaz.
Karsinojenik Ajanlar
Kimyasal karsinojenler
İnisiasyon ve promosyon olmak üzere iki basamakta etki ederler. İnisiasyonda mutasyon oluşur. Promotörlerin etkisi ile mutant hücrenin çoğalması sağlanır ve böylece transformasyon gerçekleşir. Promotörler tek başına mutajenik değildir. Önce inisiatörlerin mutasyon oluşturması gerekir. Kimyasal karsinojenler en sık RAS mutasyonuna yol açar. DNA tamir defektlerinde inisiasyon daha kolaydır ve oluşan mutasyon tamir edilemez.
İnisiatör ajanlar:
- Direkt etkililer : Beta propiolakton, Dimetil sülfat, Diepoksibütan, asetilleyici ve alkile edici antikanser ilaçlar. Kanser tedavisi sırasında lösemi, lenfoma ve diğer maliğniteler gelişebilir.
- İndirekt etkililer : p450 enzim sisteminde aktive edilirler. Metabolitleri karsinojeniktir. Fenobarbital p450 enzim sistemini indükleyerek indirekt etkili karsinojenlerin etkisini arttırır. Alkolde aynı şekilde etki edebildiğinden alkol ve sigaranın birlikte kullanılması riski tek başlarına olduklarından çok daha fazla arttırır.
• Polisiklik-heterosiklik aromatik hidrokarbonlar : Benzopiren gibi ürünler sigara dumanı, tütsülenmiş veya ateşe yakın pişirilmiş ızgara etlerde bulunurlar. Cilt, akciğer ve mesane kanseri ile sarkomlara yol açabilirler.
• Aromatik amin, amid, Azo boyaları: naftilamin , benzidin gibi ürünler karaciğerde metabolize olduğundan karaciğer ve mesane kanseri ile ilişkilidir.
• Doğal yolla oluşanlar: Aflatoksin B1, griseofulvin, safrol, sikazin vb. Aflatoksin Aspergillus flavus ‘un ürünü olup uygun koşullarda depolanmayan mısır, pirinç ve fındık gibi ürünlerde bulunur. Hepatoselüler karsinoma yol açar. HBV ile birlikte olduğunda, özellikle Çin gibi ülkelerde, risk çok artar.
• Nitrozamin ve amidler: Gıdalara koruyucu madde olarak katılan nitritler gastrointestinal sistemde dönüşüme uğrayarak özefagus ve mide kanserine yol açabilir.
• Aflatoksin, asbest, vinil klorid gibi meslek ortamında maruz kalınan kimyasal etkenlerde indirekt etkili karsinojenlerdir.
Promosyon : Forbol esterleri (TPA), hormonlar, fenoller, ilaçlar
Protein kinaz C aktivasyonu ile hücre çoğalmasını arttırırlar. İnisiasyon sonrası mutasyona uğrayan hücre çoğaldıkça maliğnite riski artar. Hormonlardan östrojen endometrial hiperplazi oluşturarak hücre çoğalmasını arttırır. DES kullanımı, benzer şekilde tümöre yol açabilir. Alkol ve safra tuzlarınında promotör etkisi olduğu ileri sürülmüştür.
Radyasyon karsinojenezi
• Ultraviole :UVB ışınları primidin dimerleri oluşturarak DNA mutasyonuna yol açar. Skuamöz (yassı epitel) hücreli ve bazal hücreli karsinom ile maliğn melanom riskini arttırır. İmmün yetmezlik varsa risk daha çok artar.
• Kseroderma pigmentozum, aşırı fotosensitivite, güneşe maruz kalan ciltte 2000 kat tümör riski artışı ve bazen nörolojik anomalilerle karakterli, OR geçişli bir DNA tamir defektidir.
• İonize radyasyon : En sık lösemi (KLL hariç) ve tiroid karsinomu (papiller) görülür. Daha az oranda meme, akciğer, tükrük bezi (mukoepidermoid karsinom) tümörleri oluşur. Cilt, gastrointestinal sistem ve kemik; radyasyona bağlı tümör gelişimine nispeten dirençlidir. Yine de, yeterli düzeyde radyasyona maruz kalan her hücre, kanser hücresine dönüşebilir. Karsinojenik etkisi dışında, iyonize radyasyon, akut dönemde, hızla çoğalan hücrelerde hasar ve ölüm oluşturur. Kemik iliği hücreleri, germ hücreleri, GİS epitel hücreleri hızla zedelenir. Periferik sinir, olgun kemik ve nöronlar nispeten dirençlidir.
Enfeksiyöz Karsinojenler
DNA virüsleri:
• HPV: Tip 16,18,31,33,35,51 serviks ve anogenital skuamöz hücreli karsinom, bazen ağız ve larinks karsinomu, Tip 6,11 (düşük risk) kondilom, Tip 1,2,4,7 skuamöz papillom (siğil). Maliğn tipleri direkt etkilidir. E6 proteini p53’e; E7 proteini Rb genine bağlanır ve inisiatör etki yapar. Sigara, mikrobial enfeksiyonlar, beslenme bozuklukları ve hormonal değişiklikler kofaaktör olarak rol oynar. İmmün yanıt da önemlidir.
• Epstein Barr virüsü : Burkitt lenfoma, B hücreli lenfomalar (öz. AİDS ve transplantasyon), bazı Hodgkin olguları, nazofaringeal karsinomlar (lenfoeepitelioma) ve immun yetmezlikte leiomyosarkom gelişimine yol açar. EBV, orofarinks ve CD21 molekülü aracılığı ile B lenfositlerini enfekte eder. Enfeksiyon latent olup lenfositi öldürmez ama immortalize edebilir. EBV, direkt karsinojenik değildir. Genetik ve çevresel faktörlerde etkilidir.
• HSV tip 8: Kaposi sarkomu, AIDS’li hastada effüzyon lenfoması (B hücreli) ve mulitiple myelomda suçlanmaktadır.
• Hepatit B virüsü: Hepatoselüler karsinom ile ilşkilidir. Uzak doğu ve Afrika’da endemiktir. Tayvan’ da HBV ile enfekte olanlarda tümör riski, olmayanlara göre 200 kat yüksektir. Tek başına direkt etkili değildir. Aflatoksin ile birlikte, direkt etki yapabilir. Virüsün HBx proteini, p53’ü etkileyerek hücre çoğalmasını arttırabilir.
RNA virüsleri
• HTLV-1 : CD4+T lenfositlerinin maliğn tümörü olan T hücreli lösemi/ lenfomayı oluşturur. Japonya ve Karayiplerde endemik; diğer bölgelerde sporadiktir. Cinsel ilişki, kan ürünleri ve emzirme ile enfekte T lenfositleri bulaşır. Enfekte olanların %3-5 kadarında, 40-60 yıl sonra tümör gelişir. Lösemi yanı sıra, tropikal spastik paraparezi denilen demyelinizan hastalık, uveit ve artrit gelişebilir.
• Hepatit C virüsü : Kronik karaciğer hasarı ve inflamasyonu izleyerek oluşan rejenerasyon, genetik instabilite ve tümör gelişimine yol açabilir.
Helikobakter Pylori
Kronik gastritli hastaların %90’ında H.pylori mevcuttur. Çoğunda klinik tablo oluşturmaz. %20-30 ülsere yol açar. Az olguda karsinom ve lenfoma oluşturabilir. CagA (sitotoksin ile ilişkili gen A) geni içeren grubu ile hastalık daha sıktır. Virulansla ilişkili VacA geni, apoptoza yol açar.
• Kronik gastrit, multifokal atrofi ve düşük asit sekresyonu, intestinal metaplazi ve displazi üzerinden, intestinal tipte gastrik karsinoma yol açar.
• H. Pylori, B hücrelerinin aktif proliferasyon göstererek t(11;18) translokasyonu gibi genetik anomaliler edindiği lenfoid infiltrasyonlara yol açar. Mukoza ile ilişkili lenfoid doku tümörleri olan MALT (marginal zon) lenfoma oluşur. H. Pylori tedavisi ile tümör gerileyebilir.
Klinik Bulgular
• Lokal ve hormonal etkiler: Hipofiz adenomu, beniğn olduğu halde, sağlam kalan hipofizide sıkıştırarak ağır endokrinopati yapabilir. Yada lümeni tıkayan bir tümör, barsakda obstrüksiyon oluşturabilir. Endokrin organ tümörlerinde, üretilen hormon artabilir. Tümörün oluşturduğu destrüktif büyüme ülserasyon, kanama ve sekonder enfeksiyonlar oluşturabilir.
• Kaşeksi: İlerleyici kilo kaybı, güçsüzlük, iştahsızlık ve anemi ile karakterlidir. En önemli etken tümör hücreleri ve makrofajlar tarafından üretilen TNF (kaşektin) dir. Ayrıca IL-1 ve INF-gama gibi sitokinler etkili olabilir.
• Paraneoplastik sendromlar: Tümörün lokal yada uzak yayılımı veya normalde o hücre tipi tarafından üretilemeyen hormon veya peptid benzeri maddelerin üretilmesi ile oluşan klinik tablolardır. En sık görülen endokrinopati Cushing sendromudur. En sık akciğer ve özellikle de küçük hücreli karsinom ile birlikte görülür. En sık görülen paraneoplastik sendrom ise hiperkalsemidir. Gri-siyah plaklar şeklinde hiperkeratotik lezyonlar ile karakterli Akantozis nigrikans, 40 yaş üzerinde görülürse, %50 maliğnite ile ilişkilidir. DİK, en sık akut promyelositik lösemi ve prostat karsinomu ile ilişkilidir.
Paraneoplastik Sendromlar:
|
Klinik sendrom |
Kanser tipi |
Mekanizma |
|
Cushing’s sendromu |
Akc.küçük hücreli ca. Pankreatik ca Neural tm. |
ACTH veya benzer maddeler |
|
Uygunsuz ADH salınımı |
Akc. Küçük hücreli ca İntrakranial neoplazmalar |
ADH veya atrial natriüretik hormonlar |
|
Hiperkalsemi |
Akc. Yassı epitel hüc. ca Meme ca Renal ca EriştinT.hüc.lösemi/lenfoma Over ca |
Parathormon ilişkili peptidler TGF-a, TNFa, IL-1 |
|
Hipoglisemi |
Fibrosarkom Diğer mezenşimal tm. Hepatosellüler ca |
İnsülin veya benzer maddeler |
|
Karsinoid send. |
Bronşial adenom(karsinoid) Pankreatik ca. Gastrik ca. |
Serotonin, bradikinin? Histamin |
|
Polisitemi |
Renal ca Serebellar hemanjiom Hepatosellüler ca. |
Eritropoetin |
|
Sinir ve kas sendromları |
||
|
Myastenya Santral, periferal sinir sist. Boz. |
Bronkojrnik ca, Timoma Meme ca |
İmmunolojik? Toksik? |
|
Dermatolojik bozukluklar |
||
|
Akantozis nigrikans Dermatomyozit |
Gastrik ca Akc. ca Uterin ca Bronkojenik, meme ca |
İmmunolojik ? EGF sekresyonu ? |
|
Kemik, eklem, yumuşak doku değişiklikleri |
||
|
Hipertrofik osteoratropati ve çomak parmak |
Bronkojenik ca |
Bilinmiyor |
|
Vasküler-hematolojik değişiklikler |
||
|
Venöz tromboz (tarusseau bulgusu) Nonbakteriel trombotik Endokardit Anemi |
Pankreas ca. Bronkojenik ca. Diğerleri Yaygın kanserler Timik tümörler |
Tümör ürünleri (müsin) Pıhtılaşmayı uyarır Hiperkoagulabilite Bilinmiyor |
|
Diğerleri |
||
|
Nefrotik sendrom |
Melanom, Akc. Ve kolon ca |
Tümör antijeni, immun kompleksler |
• Evre : Kanserin yayılım düzeyidir. Primer lezyonun büyüklüğü, bölgesel lenf bezlerine yayılımı, uzak metastaz değerlendirilir. Metastaz yapan tümörlerde en önemli prognoz faktörü evredir.
• Derece (grade): Diferansiasyon düzeyi ve mitoz sayısının değerlendirilmesidir. GradeI-IV arası derecelendirme yapılır. Anaplazik tümörler grade IV’ dür. Evre kadar olmasa da, prognozda önemlidir.
Tümör belirleyicileri
| Belirleyici (marker) | Kanserin türü |
| Hormonlar | |
| HCG | Trofoblastik tm, nonseminomatöz testiküler tümörler |
| Kalsitonin | Trioid meduller karsinomu |
| Kateşolamin ve metabolitleri | Feokromasitoma ve benzeri tümörler |
| Ektopik hormonlar | Paraneoplastik sendromlar |
| Onkofetal antijenler | |
| Alfa - fetoprotein | Karaciğer tümörleri, nonseminatöz testis germ hücreli tümörleri |
| Karsinoembriyojenik antijen | Kolon, pankreas, akciğer, mide, meme ca. |
| İzoenzimler | |
| Prostatik asid fosfataz | Prostat karsinomu |
| Nöronspesifik enolaz | Akciğer küçük hücreli karsinomu, nöroblastom |
| Spesifik proteinler | |
| İmmunglobulinler | Multiple myelom ve diğer gamapatiler |
| PSA | Prostat karsinomu |
| Musinler ve diğer glikoproteinler | |
| CA-125 | Over karsinomu |
| CA-19-9 | Kolon ve pankreas karsinomu |
| CA-15-3 | Meme karsinomu |
| Yeni moleküler belirleyiciler | |
| Gaita ve serumda p53, APC, RAS mutasyonları | Kolon kanseri |
| Gaita ve serumda p53 ve RAS mutasyonları | Pankreas karsinomu |
| Balgam ve serumda p53 ve RAS mutasyonları | Akciğer kanseri |
| İdrarda p53 mutasyonu | Mesane kanseri |
- Ayrıntılar
- Editör tarafından yazıldı.
- Kategori: Ders Notu Özeti
- Gösterim: 22103
BEYAZ HÜCRE TÜMÖRLERİ
Etyoloji ve patogenez
Kromozomal translokasyon ve onkogenler:
B lenfositlerinde daha sık
Herediter genetik faktörler:
Bloom sendromu, Fankoni anemisi ve ataksi telenjiektazi, Down sendromu, tip I Nörofibromatozis
Virüsler:
HTLV-1 :Erişkin T hücreli lösemi ve lenfoması
EBV: Burkitt lenfoma, Hodgkin lenfoma olgularının %30-40 kadarı, immünyetmezlikte görülen B hücreli lenfomalar ve nadir NK hücreli lenfomalar
KSHV/HHV-8: Sıklıkla plevral kavitede maliğn effüzyon şeklinde görülen B hücreli lenfoma
Çevresel etkenler:
Helikobakter pilori enfeksiyonu, gastrik B hücreli lenfoma
Gluten enteropatisi T hücreli intestinal lenfoma
HIV enfeksiyonunda B hücreli lenfoma ve EBV’ a bağlı lenfomalar
İatrojenik faktörler:
Radyoterapi ve bazı kemoterapi ajanları
WHO Klasifikasyonu
|
1) Prekürsör B hücre tümörleri |
4) Periferik T ve NK hücre tümörleri |
|
Prekürsör B lenfoblastik lösemi/lenfoma |
T hücre prolenfositik lösemi Büyük granüler lenfositik lösemi Mikozis fungoides/Sezary sendromu Periferal T hücreli lenfoma, nonspesifik Anaplastik büyük hücreli lenfoma Anjioimmünoblastik T hücreli lenfoma Enteropati ile ilişkili T hücreli lenfoma Pannikülit benzeri T hücreli lenfoma Hepatosplenik T hücreli lenfoma Erişkin T hücreli lösemi/lenfoma NK/T hücreli lenfoma, nazal tip NK hücreli lösemi |
|
2) Periferik B hücre tümörleri |
5) Hodgkin Lenfoma |
|
Kronik lenfositik lösemi/ küçük lenfositik lenfoma B hücre prolenfositik lösemi Lenfoplazmositik lenfoma Splenik ve nodal marjinal zon lenfoma Ekstranodal marjinal zon lenfoma Mantle hücreli lenfoma Foliküler lenfoma Marjinal zon lenfoma Hairy cell lösemi Plazmositom/plazma hücreli myelom Diffüz büyük B hücreli lenfoma Burkitt lenfoma |
Klasik tipler · Nodüler skleroz · Karışık hücreli · Lenfositten zengin · Lenfositten fakir Lenfosit baskın |
|
3) Prekürsör T hücre tümörleri |
|
|
Prekürsör T lenfoblastik lösemi/ lenfoma |
Akut lenfoblastik Lösemi/Lenfoma
%85 prekürsör B hücrelerinden oluşan, yaygın kemik iliği ve değişken periferik kan tutulumu gösteren çocukluk çağı akut lösemileridir. Prekürsör T hücreli olanlar daha az görülüp, çoğu adolesan erkeklerde timik tutulum ile karakterli lenfoma şeklindedir. Olguların çoğu 15 yaş altında olup beyaz ırkta ve erkek çocuklarında biraz daha sık görülür.
• Morfoloji: Normal doku yapısını bozan lenfoblast infiltrasyonu, bazı hücrelerde çentikli görünüm, yüksek mitoz oranı, ölen hücreli temizlemek için gelen makrofajlarla oluşan yıldızlı gök manzarası benzeri görünüm
• İmmünfenotip: pre-B ve pre-T hücrelerinde bulunan TdT, >%95 olguda bulunur. T ve B hücre ayırımı için spesifik belirleyiciler kullanılır. Pre-B hücresi CD19, CD10; pre-T hücresi CD1, CD2, CD5 ve CD7.
• Kötü prognoz bulguları: 2 yaş altı; adolesan ve erişkin yaş; periferik kanda 100,000 üzerinde blastik hücre; çocukluk çağında %3, erişkinde %25 olguda belirlenen t(9;22) – Philadelphia kromozomu
• İyi prognoz bulguları: 2-10 yaş; düşük sayıda hücre, erken pre-B fenotipi, hiperploidi yada t(12;21)
PERİFERİK B HÜCRE TÜMÖRLERİ
Kronik lenfositik lösemi (CLL)/ Küçük lenfositik lenfoma (SLL)
Bu iki hastalık morfolojik, fenotipik ve genotipik olarak birbirinden ayrılamaz. Aralarındaki tek farklılık, periferik kandaki lenfositoz derecesidir. Çoğu hastada lenfosit sayımı >4000/mm3 olup lösemi tanısı alır. Batılı ülkelerde en sık görülen erişkin çağ lösemisidir.
• Morfoloji: Lenf nodunu diffüz infilte eden yuvarlak, hafif düzensiz nüveli, dar sitoplazmali küçük lenfositler; lösemik formunda kanda bulunan lenfositler frajil olup yayma hazırlanırken ezilmiş hücreler
• İmmünfenotip: CD19, CD20, CD23 ve CD5 düşük düzeyde yüzey immünglobülin ekspresyonu (IgM/ IgM ve IgD)
• Klinik: Çoğu 50 yaş üzeri (median yaş 60) erkek hastalardır (M/F: 2/1). Çoğu asemptomatiktir. Jeneralize lenfadenopati, hepatosplenomegali %50-60 olguda mevcuttur. Kemik iliği tutulumu olan SLL olgularında lökopeni; ağır CLL olgularında >200,000/mm3 üzeri periferik lökositoz olabilir. Hipogamaglobülinemi sıktır. %10-15 olguda otoimmün hemolitik anemi yada trombositopeni; ortalama sağkalım 4-6 yıl; %10 olguda diffüz büyük B hücreli lenfomaya transformasyon (Richter sendromu)
Foliküler Lenfoma
ABD’ de en sık görülen NHL olup erişkin lenfomalarının %45’ ini oluşturur. En sık orta yaşta; kadın ve erkekde aynı oranda görülür. Neoplastik hücreler normal germinal merkez B hücrelerine benzer.
• Morfoloji: Lenf nodunda nodüler patern; çoğunda küçük çentikli hücreler baskın; %85 olguda kemik iliği tutulumu; lösemik dönüşüm nadir
• İmmünfenotip: CD19, CD20, CD10, yüzey immünglobülinleri BCL2 protein ekspresyonu (normal foliküler hücrelerinden farkı)
• Sitogenetik ve moleküler genetik: %90 olguda t(14;18) sonucu IgH (ağır zincir) lokusu ile 18. kromozomdaki BCL2 lokusu birleşir ve BCL2 ekspresyonu artar. Apoptoz engellenir ve neoplastik B hücreleri çoğalır.
• Klinik: Ağrısız, jeneralize lenfadenopati; ortalama sağkalım 7-9 yıl; tedaviye yanıt yok; %30-50 olguda diffüz büyük B hücrei lenfomaya transformasyon
Diffüz Büyük B Hücreli Lenfoma
Tüm NHL olgularının %20; agresif lenfomaların %60-70 kadarını oluşturur. En sık 60 yaş civarında ve erkeklerde biraz daha sık görülür. Çocukluk çağı lenfomalarının %5 kadarını oluşturur.
• Morfoloji: Büyük boyutlu lenfositler ve diffüz büyüme paterni
• İmmünfenotip: CD19, CD20, bazılarında CD10 ve BCL6, çoğunda yüzey Ig
• Onkogenik virüsler ile ilişkili subtipler: Ağır T hücre yetmezliğinde (ileri evre HIV enfeksiyonu, ağır kombine immün yetmezlik, transplantasyon) EBV etkisi; ileri evre HIV enfeksiyonunda görülen vücut kavite lenfomalarında KSHV/HHV8 etkisi
• Klinik ve prognoz: Tek bir nodal yada ekstranodal bölgede, hızla büyüyen semptomatik kitle; kemik iliği geç dönemde tutulur ve lösemik tablo nadir; tedavi edilmezse kötü prognoz; yoğun kombine kemoterapi ile %60-80 komplet remisyon ve yaklaşık %50 olguda iyileşme
Burkitt Lenfoma
• Morfoloji: Orta boyutlu, birkaç nükleolü olan nüveli hücreler; sık mitoz ve apoptoz; kalıntıları fagosite eden makrofajlar nedeni ile yıldızlı gök manzarası
• İmmünfenotip: Yüzey IgM, monotipik hafif zincirler, CD19, CD20, CD10 ve BCL6 gibi matür B hücre belirleyicileri
• Sitogenetik ve moleküler genetik: Tümünde 8. kromozom da yerleşen c-MYC geninde translokasyon t(8;14).
• Endemik tümörlerin tümünde; AIDS hastalarının %25’i, sporadik olguların %15-20’ sinde latent EBV enfeksiyonu
• Klinik: Endemik ve sporadik olguların çoğu çocuk ve genç erişkinlerde görülür. ABD’ de çocukluk çağı lenfomalarının %30’ u Burkitt lenfomadır. Çoğu ekstranodal yerleşir. Endemik olanlar mandibula yanı sıra böbrek, over ve adrenal gland gibi abdominal organlarda yerleşir. Sporadik olgular daha sık olarak ileoçekal bölge ve peritonda yerleşir. Kemik iliği ve periferik kan tutulumu nadirdir. Çok agresif olmakla birlikte, kısa süreli, yüksek doz kemoterapiye iyi yanıt verir. Çoğu çocuk ve genç erişkin iyileşirken, yaşlılarda kötü davranış gösterir.
*************Çocuklarda görülen non-Hodgkin lenfomalar agresif tümörler olup kötü prognozludur. Ama hücreler hızlı çoğaldığından kemoterapiye iyi yanıt verirler ve erken tanı ile iyileşme şansları yüksektir. Burkitt lenfoma vücutta en hızlı çoğalan tümör olarak bildirilmektedir***********************
PLAZMA HÜCRE TÜMÖRLERİ
• Multiple myelom (plazma hücre myelomu) En önemli ve en sık semptomatik monoklonal gamopatidir. İskelet sisteminde yaygın odaklar yapar. Nadir görülen soliter myelom yada plazmositom ise kemik yada bazı yumuşak dokularda tek (soliter) kitle yapar.
• Waldenström makroglobülinemisi: IgM düzeyi yüksekiliğine bağlı gelişen hipervizkozite vardır. Lenfoplazmositik lenfoması olan erişkinlerde görülür.
• Ağır zincir hastalığı: Serbest H zincirleri üretip salgılayan bir gruptur. CLL/SLL, lenfoplazmositik lenfoma ve ince barsakta nadir görülen bir tümöral proliferasyon (Akdeniz lenfoması) bu tabloya yol açabilir.
• Primer yada immünosit ile ilişkili amiloidoz: serbest L zincirleri üreten plazma hücre proliferasyonu olup amiloidoz ile sonuçlanır. Bazı hastalarda belirgin multiple myelom; bazılarında ise yalnızca kemik iliğinde minör klonal plazma hücre proliferasyonu belirlenir.
• Önemi belirlenemeyen monoklonal gamopati: Klinik bulgusu olamayan hastalarda kanda m komponentlerinin belirlenmesidir. Yaşlılarda oldukça sık olmakla birlikte semptomatik olgulara (en sık multiple myelom) dönüşüm nadirdir.
Multiple Myelom
• İskelet sisteminde plazma hücrelerinden oluşan multifokal, destrüktif tümör odakları; sıklık sırasına göre vertebra, kotsalar, kafa tası, pelvis, femur, klavikula ve skapula; radyolojik olarak 1-4 cm çapında, zımba ile delinmiş görünümde lezyonlar
• Düzensiz immünglobülin üretimine bağlı hücre içi birikimler değişik görünümlere yol açar: alev kırmızısı sitoplazma (alev hücreleri); mavi görünümde üzüm tanesi benzeri sitoplazmik damlacıklar (Mott hücreleri); sitoplazmik Russel cisimcikleri; intranükleer Dutcher cisimcikleri;
• Yüksek düzeyde M proteini nedeni ile eritrositlerin birbirine yapışması: rulo formasyonu
• Enfeksiyonlardan sonra, en sık ölüm sebebi böbrek yetmezliği
• %6-15 hastada amiloidoz
• Kesin tanı kemik iliği incelemesi ile
PERİFERİK T VE NK HÜCRE TÜMÖRLERİ
Erişkin T Hücreli Lösemi/Lenfoma
• CD4+ T lenfositlerinin tümörü olup HTLV-1 virüsü ile ilişkili
• Japonya, batı Afrika ve Karaib adaları gibi, virüsün endemik olarak bulunduğu yerlerde daha sık
• Cilt lezyonları, jeneralize lenfadenopati, hepatosplenomegali, periferik lenfositoz ve hiperkalsemi
• Tutulan bölgeler ve periferik kanda multilobe nüveli (çiçek hücreleri) hücreler
Mikozis Fungoides/Sezary Sendromu
• CD4+ T lenfositlerinin maliğn tümörü olup cilt tutulumu ön planda
• Epidermis ve üst dermisi infiltre eden maliğn hücrelerda serebriform nüveler
• Ekstrakutanöz yayılım lenf nodu ve kemik iliğine
• Sezary sendromunda jeneralize eksfoliatif eritrodermi ve eşlik eden Sezary (serebriform) hücrelerini içeren lösemi
• Yavaş gidişli olup ortalama sağkalım 8-9 yıl
************Non-Hodgkin lenfomaların bir kısmında lösemik dönüşüm sıktır. Bu grupta lösemi ve lenfoma hücreleri aynı genotipik, fenotipik ve morfolojik özelliklere sahiptir. Bu tümörler: küçük hücreli lenfoma/kronik lenfositik lösemi; T hücreli lenfoblastik lenfoma/T hücreli ALL; mikozis fungoides/Sezary sendromu****************
HODGKİN LENFOMA
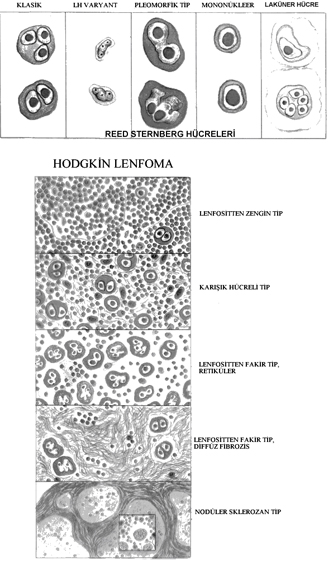
Primer olarak lenfoid dokuları tutan bir hastalıktır. Tek bir lenf nodu veya nod zincirinde gelişip
ilişkili olduğu lenf nodlarına yayılır.
• Reed-Sternberg (RS) denilen neoplastik dev hücreler
• Ateş gibi belirgin klinik bulgular ile NHL’dan ayrılır
Reed- Sternberg hücreleri germinal merkez yada post-germinal merkez B hücrelerinden köken alırlar. Bu nedenle Hodgkin lenfoma B hücre tümörü olarak kabul edilmektedir. Genç erişkinlerde en sık görülen maliğn tümörlerden biri olup tanı anında ortalama yaş 32 dir. Tedavide gelişmeler sonucunda çoğu olguda iyileşme sağlanmaktadır.
WHO sınıflaması:
• Nodüler sklerozis
• Karışık hücreli
• Lenfositten zengin
• Lenfositten fakir
• Lenfosit baskın
***************İlk dört tipinde (klasik tipler) Reed-Sternberg hücrelerinde CD20 negatif; CD15 ve CD30 pozitiftir. Lenfosit baskın tipte lenfohistiositik varyant (popkorn) hücrelerde CD20 pozitif; CD15 ve CD30 negatiftir.***************************
• Nodüler sklerozis tipi: En sık görülen tip (%65-70). Laküner hücreler ve lenf nodunu nodüllere ayıran kollajen bantlar görülür. F=M. Alt servikal, supraklaviküler ve mediastinal lenf nodu tutulumu sıktır. Ergenlik ve genç erişkinlerde sık olup nadiren EBV ile ilişkilidir. Prognozu mükemmeldir.
• Karışık hücreli tip: %20-25 olguyu oluşturur. Küçük T lenfositi, eozinofil, plazma hücresi ve beniğn makrofajlardan oluşan hücrelerin arasında diagnostik ve mononükleer Reed-Sternberg hücreleri görülür. Erkeklerde daha sıktır. %70 kadar olguda EBV ile ilişkilidir. İleri yaşta, sistemik semptomlarla birlikte olup sıklıkla ileri evrededir. Yine de prognoz çok iyidir.
• Lenfositten zengin tip: Nadirdir. Mononükleer ve diagnostik Reed-sternberg hücrelerinin sık olması ile lenfosit baskın tipten ayrılır. %40 olguda EBV ile ilişkilidir. Prognozu çok iyi yada mükemmeldir.
• Lenfositten fakir tip: En az görülen tiptir. Lenfositler az olup Reed-Sternberg hücreleri ve pleomorfik tipleri sıktır. Büyük hücreli NHL ile karışır. Yaşlılarda daha sık görülür. HIV (+) kişilerde, geri kalmış toplumlarda daha sık olup sıklıkla EBV ile ilişkilidir. İleri evre ve sistemik bulgular sık olup diğer tiplere göre kötü prognozludur.
• Lenfosit baskın tip: Nadirdir. Olguların %5 kadarını oluşturur. Beniğn histiositler ile karışık küçük lenfosit infiltrasyonu görülür. Tipik Reed- Sternberg hücreleri azdır. Lenfohistiositik varyant (popkorn hücre) sıktır. Eozinofil, plazma hücresi ve nötrofiller az olup nekroz ve fibrozis izlenmez. EBV ile ilişkili değildir. Hastaların çoğu 35 yaş altı, erkeklerdir. Servikal yada aksiller lenfadenopati sık olup mediasten yada kemik iliği tutulumu nadirdir. Tekrarlama riski olsa da, prognoz mükemmeldir.
LANGERHANS HÜCRELİ HİSTİOSİTOZ
Dendritik yapıdaki Langerhans hücreleri HLA-DR, S-100 ve CD1a eksprese ederler. Sitoplazmada Birbeck granülleri karakteristiktir.
Multifokal multisistem Langerhans hücreli histiositoz (Letterer-Siwe hastalığı)
• En sık 2 yaş altında
• Seborreik döküntülere benzeyen cilt lezyonları, hepatosplenomegali, lenfadenopati, pulmoner lezyonlar ve sonunda destrüktif kemik lezyonları
• Kemik iliği infiltrasyonu pansitopeni, tekrarlayan otitis media ve mastoidit gibi enfeksiyonlar
• Tedavi edilmez ise öldürücüdür. Yoğun kemoterapi ile 5 yıllık sağkalım %50’ dir.
Unifokal unisistem Langerhans hücreli histiositoz (eozinofilik granülom)
• En sık kemik iliğini tutan, genişleyen eroziv lezyonlar
• Histiositler eozinofil, nötrofil, lenfosit ve plazma hücreleri ile karışık
• En sık kafa kemikleri, kotsalar ve femurda
• Asemptomatik olabilir yada ağrı, hassasiyet ve patolojik kırığa yol açar.
• Spontan olarak gerileyebileceği gibi, lokal eksizyon yada radyoterapi ile iyileşir.
•
Multifokal unisistem Langerhans hücreli histiositoz
• Genellikle küçük çocuklarda, multifokal, yumuşak dokuya uzanan, eroziv kemik defektleri
• Hastaların yaklaşık %50’sinde arka hipofiz sapı tutulumu sonucunda diabetes insipitus
• Kalvarial kemik defektleri, diabetes insipitus ve ekzoftalmus kombinasyonuna Hand-Schüller-Christian triadı denir.
• Çoğu hastada spontan gerileme;diğerlerinde kemoterapi ile tedavi
TİMUS
Hiperplazi : Normal timusta lenfoid foliküller yoktur. Hiperplazide medullada lenfoid foliküller oluşur. Myastenya gravis, SLE ve romatoid artrit gibi otoimmun hastalıklarda çoğunlukla timusda hiperplazi oluşur. Myastenya graviste timusun myoid hücrelerine karşı duyarlılık kazanan T hücreleri lenfoid foliküllerde B hücrelerini uyararak antikor oluşumuna yol açar. Bunun sonucunda nöromuskuler bileşkedeki asetil kolin reseptörlerine karşı otoimmun reaksiyon gelişir.
Timoma
Timustaki epitelyal hücrelerin tümörleridir. Tümörde bulunan lenfositler normal yapıda olup neoplazik değildir.
Beniğn Timoma : Sitolojik ve biolojik olarak beniğndir. Medulladaki epitel hücrelerine benzer hücreler oluşturur. Timomaların %60-70’ini oluşturur.
Malign Timoma : Tip I : Sitolojik olarak beniğn görünsede lokal invazyon ve nadiren uzak metastaz yapabilir. Timomaların %20-25’inin oluşturur.
Tip II (Timik Karsinom) :Hem sitolojik olarak hemde biolojik olarak maligndir. Timomaların %5’ini oluşturur. Çoğu iyi veya az diferansiye skuamöz karsinomdur. Daha az olarak görülen lenfoepitelyoma da olgun görünümlü lenfositlerde tümöre eşlik eder. Bir kısmında EBV varlığı belirlenmiştir. Bu açıdan nazofaringeal karsinoma benzer.
Timomalar orta yaşta sıktır. Myastenya gravisli olguların %15-20’sinde timoma bulunur. Tümör çıkarıldığında nöromuskuler bozukluk geriler. Hipogamaglobulinemi, SLE, saf eritrosit aplazisi ve nontimik kanserlerde timoma ile ilişkili olabilir.
DALAK
Primer tümörleri fibrom, osteom, kondrom ve en sık görülen lenfanjiom ile hemanjiom; maliğn olarak anjiosarkom, fibrosarkom.
Splenomegali :
Çoğu olguda splenomegali başka bir bölgedeki primer hastalığa sekonder olarak gelişir. Eritrosit, trombosit ve lökositler dalakta yıkılabilir.
İleri Derecede Splenomegali (1000 gm )
1. Kronik Myeloproliferatif hastalık (KML, Myeloid metaplazi)
2. KLL (daha az oranda)
3. Hairy cell lösemi
4. Sıtma
5. Gaucher hastalığı
6. Lenfomalar
7. Primer dalak tümörleri (nadir)
Orta Derecede Splenomegali (500-1000 gm)
1. Kronik konjestif splenomegali (portal hipertansiyon veya splenik ven obstrüksiyonu)
2. Akut lösemi
3. Herediter Sferositoz
4. Talasemi mayor
5. Otoimmun hemolitik anemi
6. Amiloidoz
7. Niemann-Pick hastalığı
8. Langerhans hücreli histiositoz
9. Kronik splenit (özellikle enfektif endokardit ile birlikte)
10. Tüberküloz, sarkoidoz, tifo
11. Metastatik karsinom veya sarkom
Hafif derecede Splenomegali ( 500 gr. altında)
1. Akut splenit
2. Akut splenik konjesyon
3. Enfeksiyöz mononükleoz
4. Septisemi, SLE, intra-abdominal enfeksiyonlar gibi akut febril hastalıklar.
Dalak büyümelerinde hipersplenizm oluşabilir anemi, lökopeni, trombositopeni
****************Anemi ve splenomegali ilişkisi sık olarak soruluyor. Herediter sferositoz, otoimmün hemolitik anemi ve talasemi major dışında kalan anemilerde belirgin splenomegali görülmez.**********